1. Framework
1.1 Overall framework of Dr. Kinase
Protein kinases (PKs) regulate various cellular functions and hold significant pharmacological promise in cancer and other diseases. As of September 2024, the FDA has approved 82 inhibitors that primarily target different kinases. However, PKs targeted by small-molecule inhibitors develop resistance through mutations in four types of representative mutation hotspots (gatekeeper, G-loop, αC-helix, and A-loop). One notable example is the T315I gatekeeper mutation in ABL1, which is resistant to imatinib, dasatinib, and nilotinib. In the past decade, kinase inhibitor (KI) drug-resistance has become a common clinical complication affecting multiple cancers, targeted kinases, and drugs. To tackle this challenge, we built a timely web service, named Dr. Kinase, for predicting the loci of four types of drug-resistance hotspots in protein kinases.
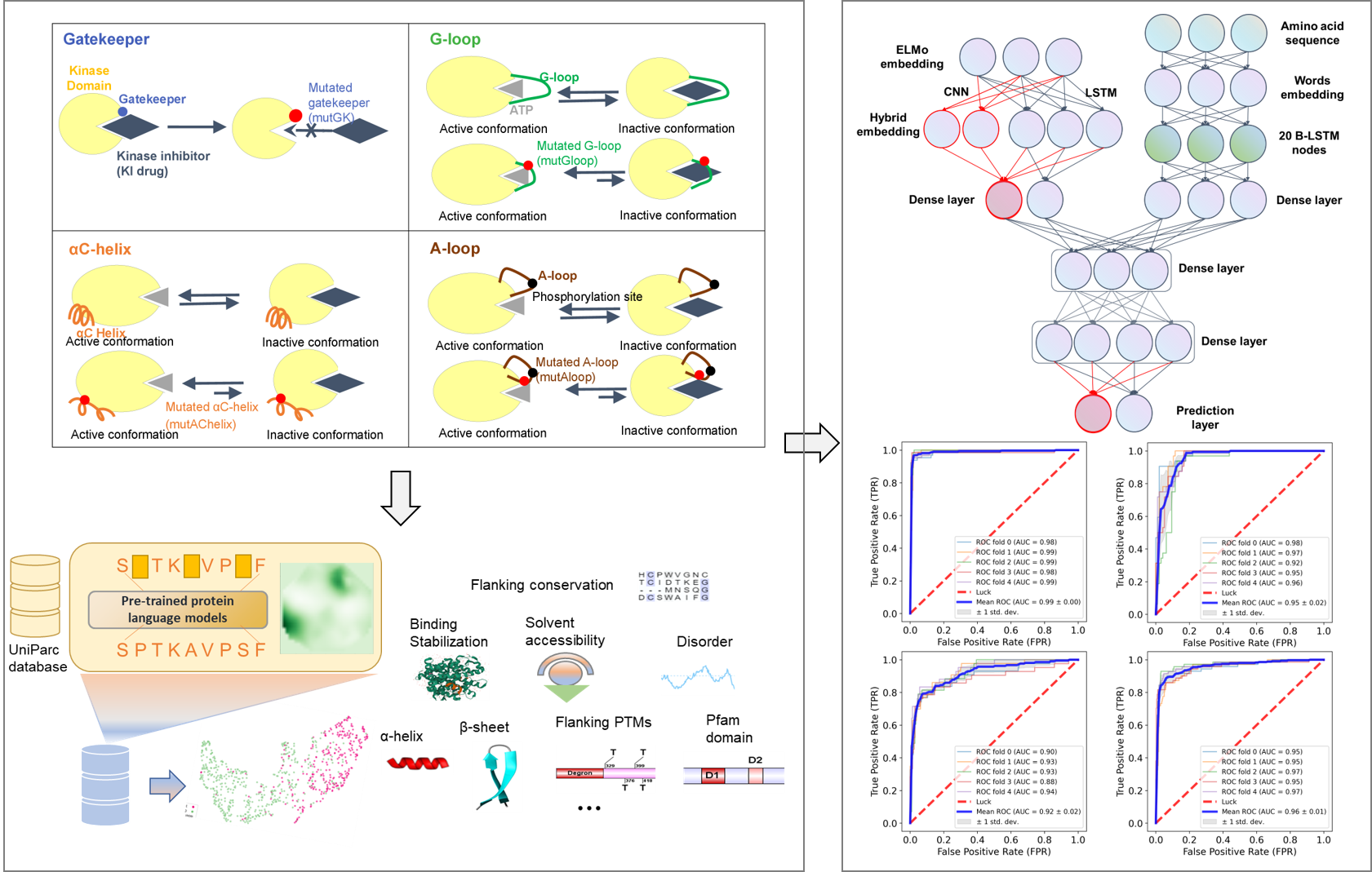
Dr. Kinase builds upon our previously published studies (Hu R et al. NAR, 2021; Kim P et al. BIB, 2021) and utilizes the advantages of deep hybrid learning technology and multimodal features to predict actionable drug-resistance hotspots (Figure 1). The performance of Dr. Kinase has been rigorously evaluated using five-fold cross-validation and additional independent testing, demonstrating excellent accuracy with area under the curve (AUC) values exceeding 0.89 in different types of drug-resistance hotspot predictions. Additionally, Dr. Kinase provides comprehensive annotations and visualizations for the predicted results. This server is essential not only for its capacity to unveil the potential underlying mechanisms of KI drug resistance but also to aid the cancer and drug research communities in the development of next-generation KIs for emerging cancer precision medicine. Through Dr. Kinase, we hope to serve users in broad fields, including basic, translational and clinical research.
1.2 Collection and process of datasets
To date, there are four four types of representative mutation hotspots (gatekeeper, G-loop, αC-helix, and A-loop) as following:
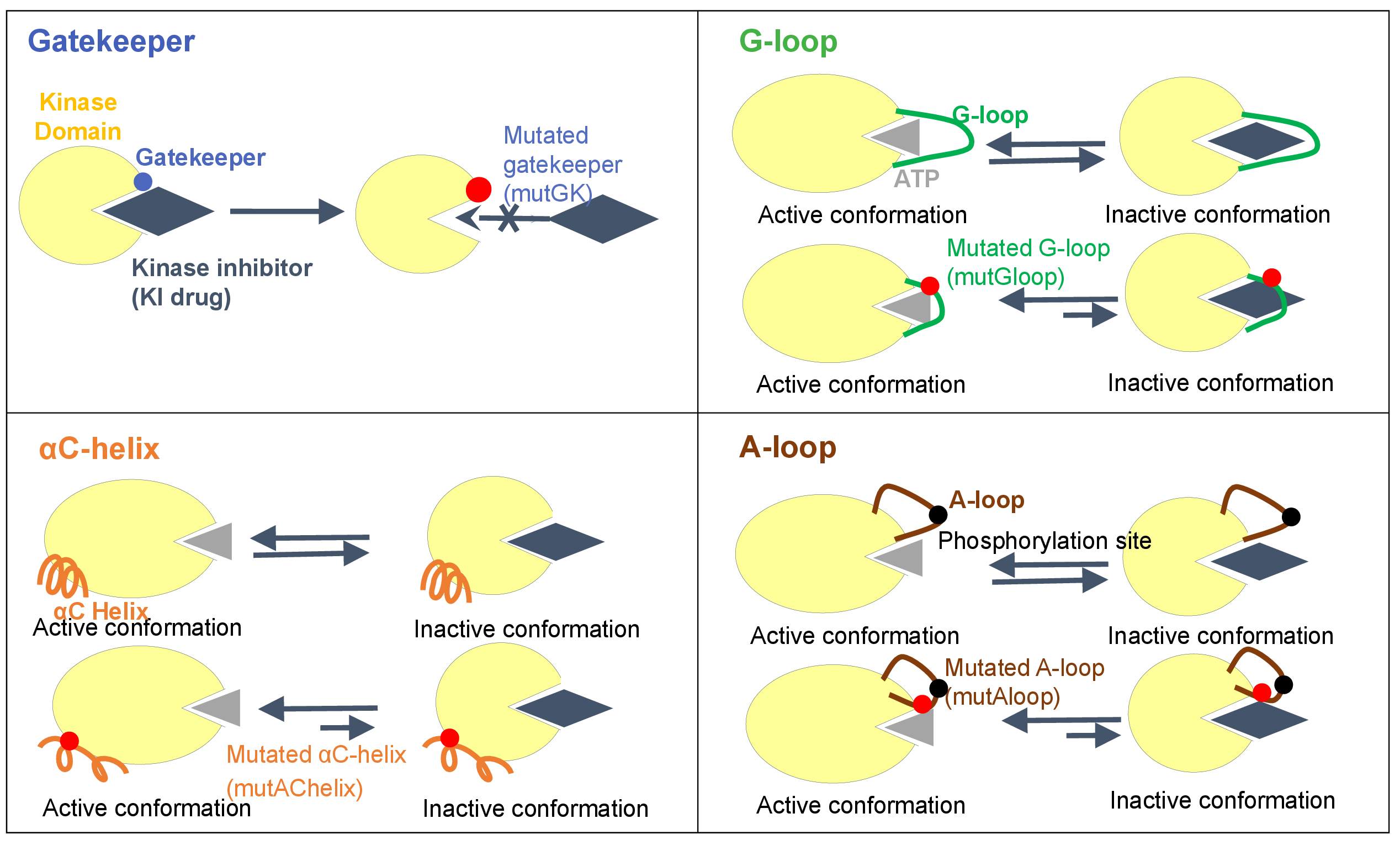
- Gatekeeper residue is a single amino acid located near the protein-drug binding site. In wild-type kinases, gatekeeper residues have small side chains that sterically accommodate drugs. These small side chains can be mutated into bulky side chains that impede drug-protein binding. This confers drug resistance.
- A-loop, which called activation loop or T-loop, is located in the C-lobe. A-loop contains a phosphorylation site, which upon phosphorylation, induces a conformational change on the loop, and then allows a substrate to bind. The activation loop and P+1 loop constitute the activation segment that runs from the DFG (Asp-Phe-Gly) motif to the APE (Ala-Pro-Glu) motif. A-loop mutations may have indirect effects that disfavor drug binding by increasing entropy or destabilizing the inactive conformation.
- G-loop, which is also named glycine-rich loop, and P-loop (phosphorylation loop) have a conserved consensus motif GxGxxG, where G represents glycine, and x can be any amino acid. G-loop mutation can cause clinical resistance to type 2 kinase inhibitors (T2KIs) by destabilizing the inactive conformation, stabilizing the active conformation, and/or removing direct drug interactions.
- αC-helix, also called C-helix and αC, is a single α-helix located in the N-lobe of KD between β3 and β4 strands. αC-helix is usually the first α-helix in the kinases domain (from N-terminus), but in some kinases such as AGC kinases, a short αB–helix may precede αC-helix.
We obtained the substructure locus information of 547 human kinases from our KinaseMD databases. After removing duplicate kinases and filtering out the kinases without specific locus information of four categories mentioned above, we obtained 388 unique human kinases. Among them, 344, 312, 172, and 231 kinases had Gatekeeper, A-loop, G-loop, and αC-helix locus information, respectively.
1.3 Investigations of drug resistance hotspots
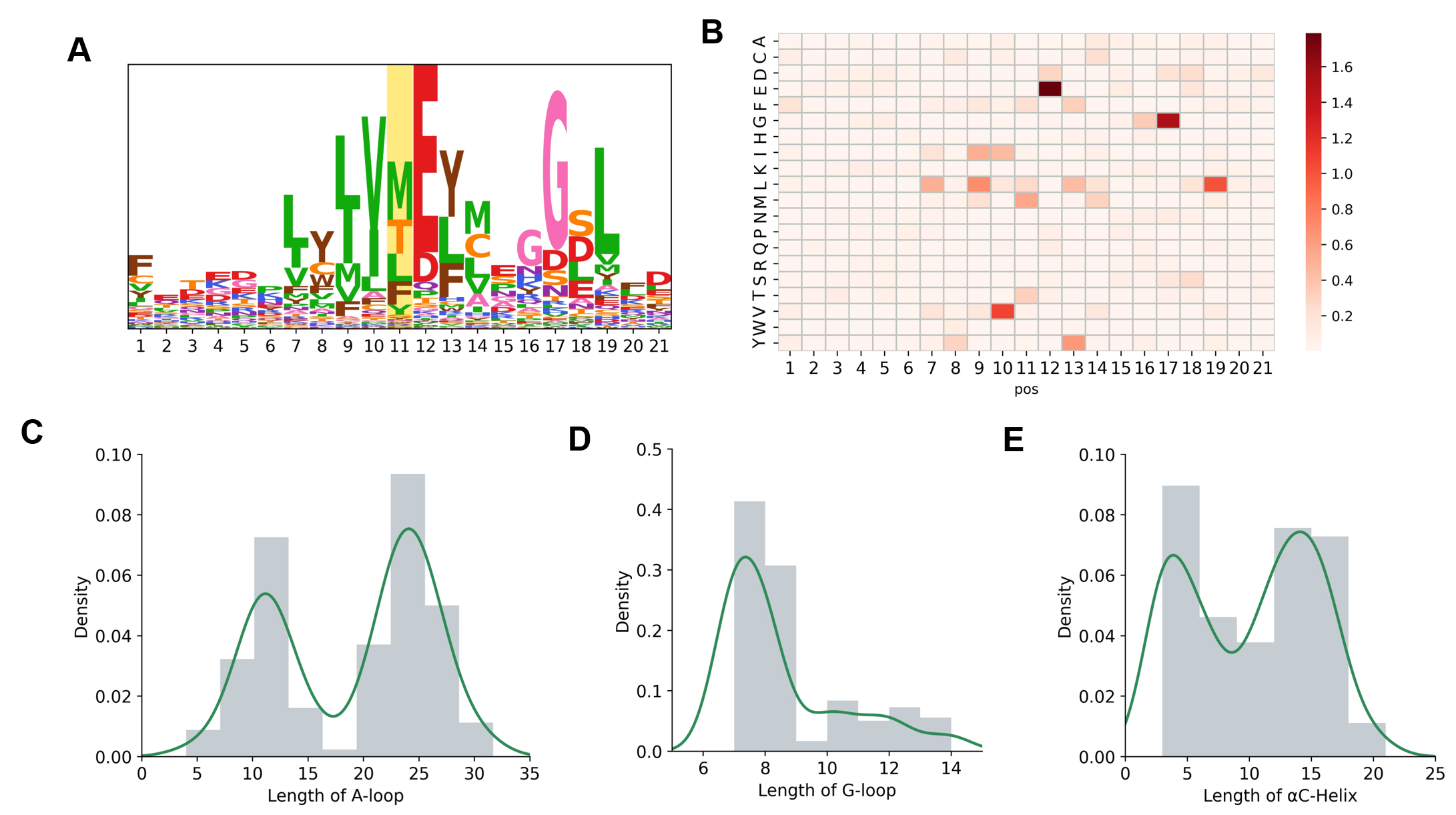
Based on the collected information on drug resistance hotspots, we analyzed the amino acid preference and length of different hotspots. More specifically, there are significantly more Methionine (M), Threonine (T), Leucine (L), and Phenylalanine (F) residues on gatekeeper. In addition, we found that L, Glutamate (E), Valine (V), Isoleucine (I), and Glycine (G) were significantly enriched upstream and downstream of gatekeeper. In addition, for A-loop, G-loop, and αC-helix, we count their most likely sequence lengths. For example, the most representative lengths of A-loop are 24 and 12, which is also the length of the peptide library they cut into when making their predictions.
1.4 Characteristics of drug resistance hotspots
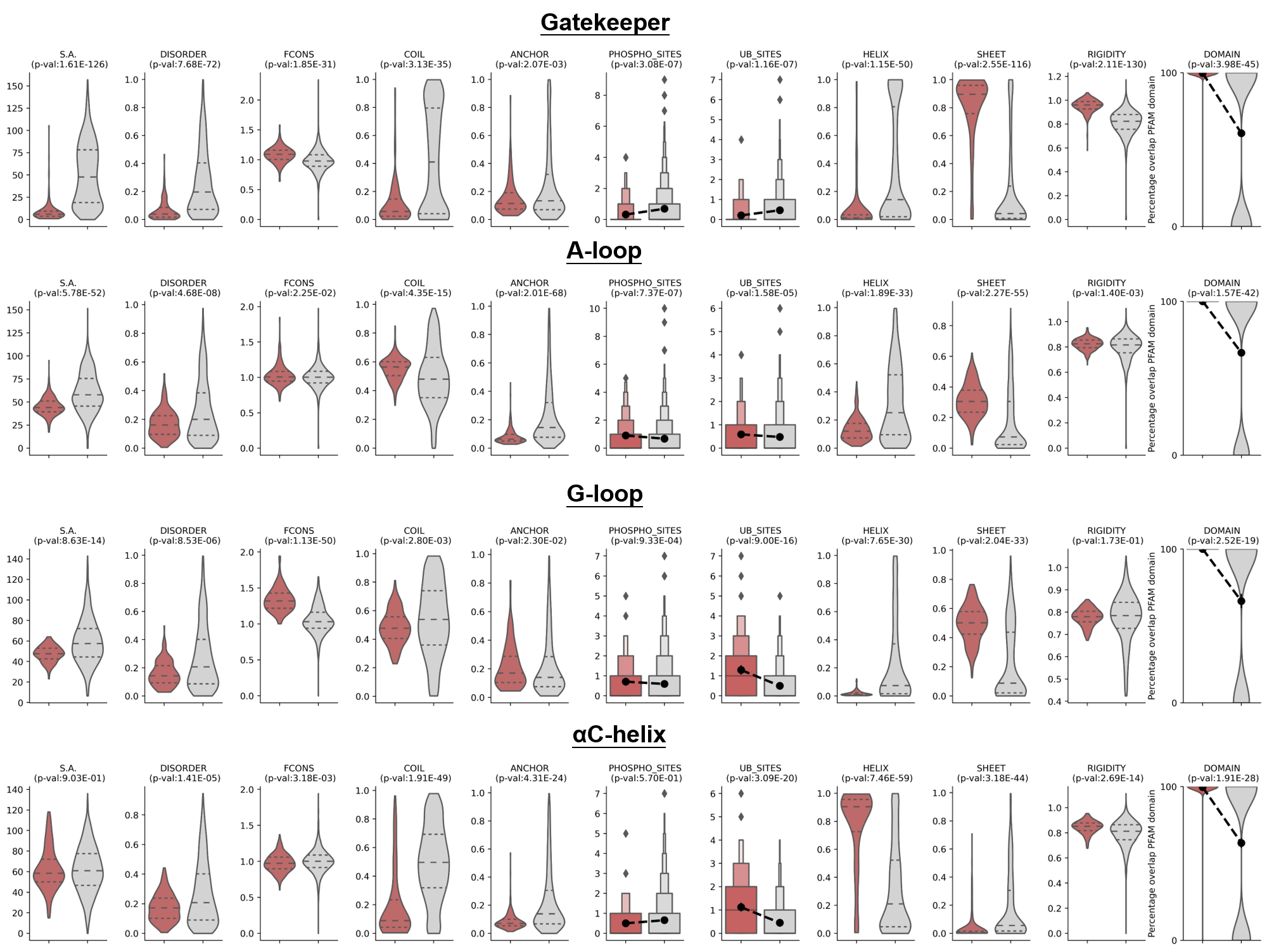
Understanding the structure characteristics of drug resistance hotspots is crucial for predicting their resistance potential and unraveling their functional implications. To characterize the structure characteristics of drug resistance hotspots, we analyzed the curated, experimentally validated hits by comparing to the background dataset comprising a considerable number of randomly selected peptides with the same length as the drug resistance hotspots. We employed various structural bioinformatics algorithms and tools to identify common structural properties.
Gatekeeper and its surrounding sequences were more evolutionarily conserved and enriched in functional domains. Remarkably, the known gatekeepers exhibited a lower degree of solvent accessibility and protein disorder compared to the random peptides. Furthermore, known gatekeepers were found to be preferentially located in higher flexibility regions and had higher binding stability. Additionally, the analysis revealed a specific preference of Gatekeeper for coiled coil and sheet regions rather than α-helix regions. Sequences of A/G loops were more evolutionarily conservative and enriched in functional domains as well. On the contrary, they were found to be preferentially located in protein disordered regions, highlighting their distinctive localization patterns. For αC-helix, the analysis revealed a specific preference of α-helix regions rather than coiled coil and sheet regions. It was also observed that αC-helix tends to occur in lower flexibility regions. These findings provided valuable insights into the structural characteristics of drug resistance hotspots and indicate potential determinants for their recognition.
1.5 Deep hybrid learning technology for predicting actionable drug-resistance hotspots
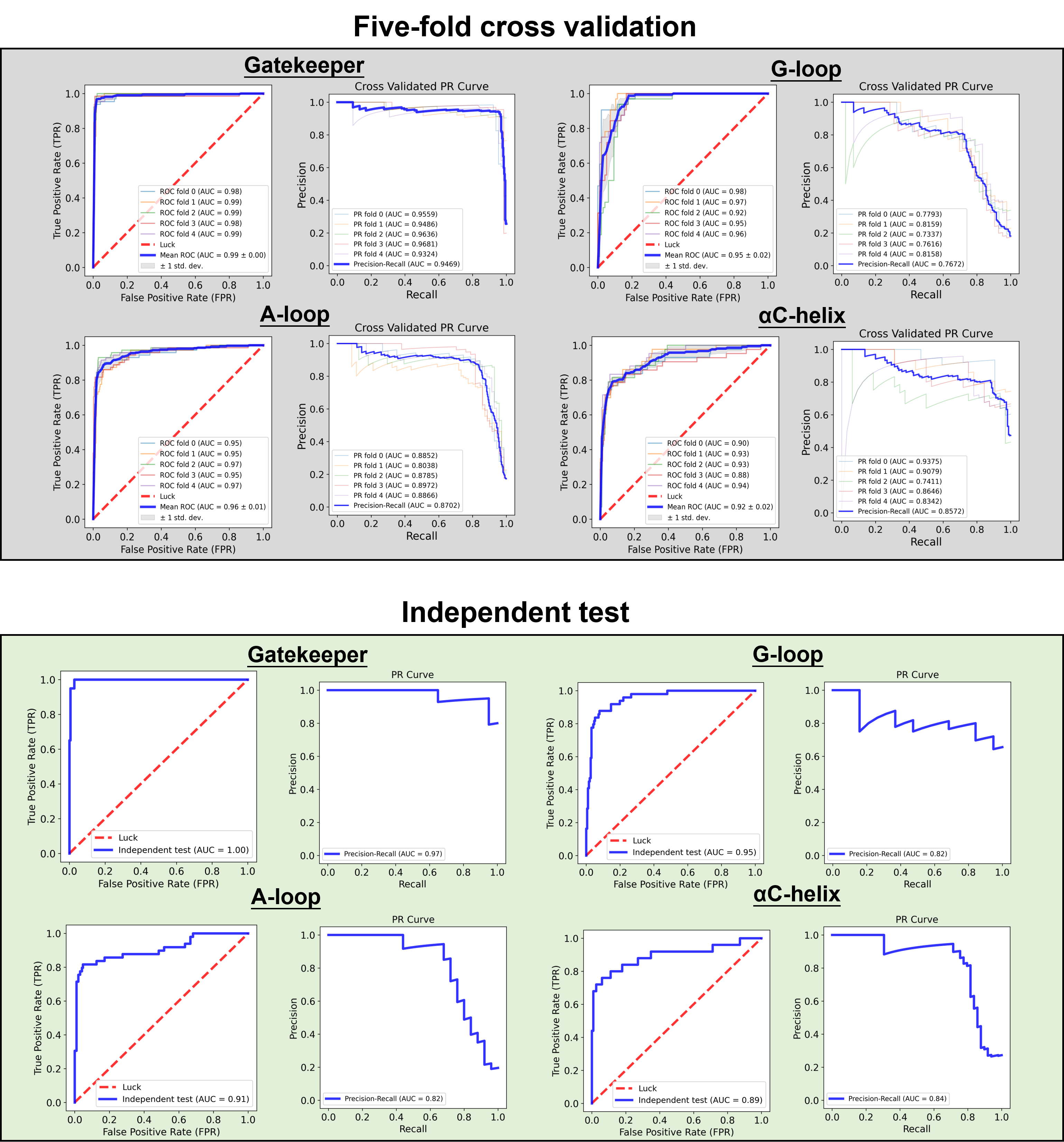
Dr. Kinase utilized a hybrid architecture comprising cutting-edge deep learning networks, such as protein language models, word embeddings, convolution, and Bidirectional Long Short-Term Memory (BLSTM). This deep learning framework integrating sequential, evolutionary, and structural features allows Dr. Kinase to leverage the full potential of these advanced networks and their ability to extract high-level features from protein sequences. The performance evaluation of Dr. Kinase demonstrated its great predictive capabilities. Through a five-fold CV approach, Dr. Kinase obtained an average AUC value of 0.96 in predicting four types of drug resistance hotspots ranging from 0.92 to 0.99, indicating consistent and reliable performance (gatekeeper: 0.99, G-loop: 0.95, A-loop: 0.96, αC-helix: 0.92). Additionally, when tested with an independent dataset, Dr. Kinase achieved an average AUC value of 0.94 among different drug resistance hotspots (gatekeeper: 1, G-loop: 0.95, A-loop: 0.91, αC-helix: 0.89). These results demonstrate the robustness and accuracy of Dr. Kinase in predicting drug resistance hotspots in protein kinases.
2. Prediction
Protein kinases have been prominent targets for modern drug discovery against many diseases. As of September 2024, the FDA has approved 82 inhibitors targeting different kinases. However, PKs targeted by small-molecule inhibitors develop resistance through mutations in four types of representative mutation hotspots (gatekeeper, G-loop, αC-helix, and A-loop). Dr. Kinase represents a unique prediction server designed specifically for predicting the loci of four drug-resistance (DR) hotspots in protein kinases.
2.1 Inputs
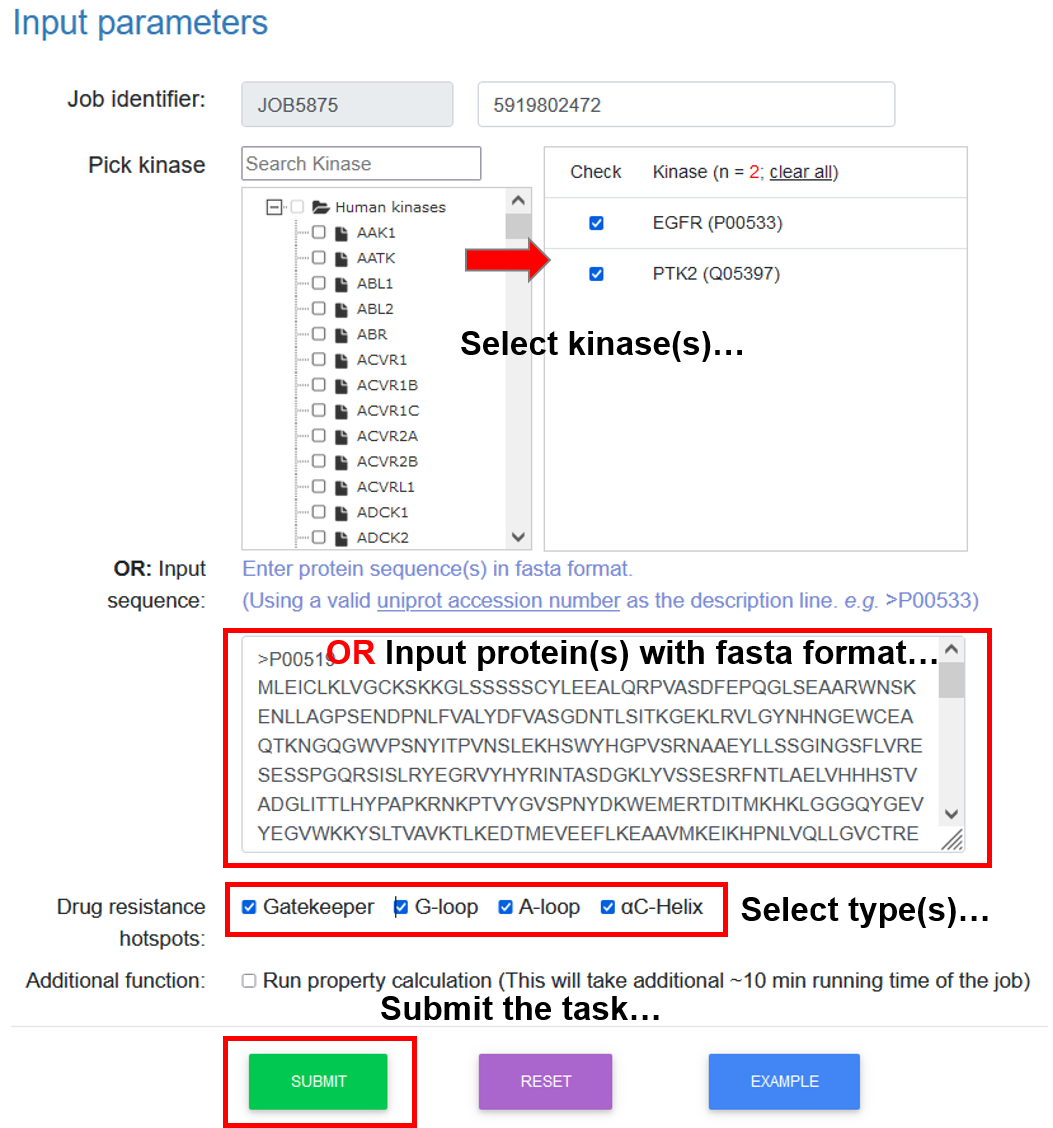
Input: 1) Select or enter kinase protein sequence(s) in FASTA format; 2) Select hotspot type(s) (gatekeeper, G-loop, αC-helix, and A-loop).
3. Result and visualization
Output: 1) Predicted DR loci of selective type(s) and visualization in sequence and 3D structure; 2) Calculated structural and physicochemical features for predicted DR loci; 3) Visualizations of cancer mutation map, kinase substrate network, and MSA in species, etc.
3.1 Basic prediction information
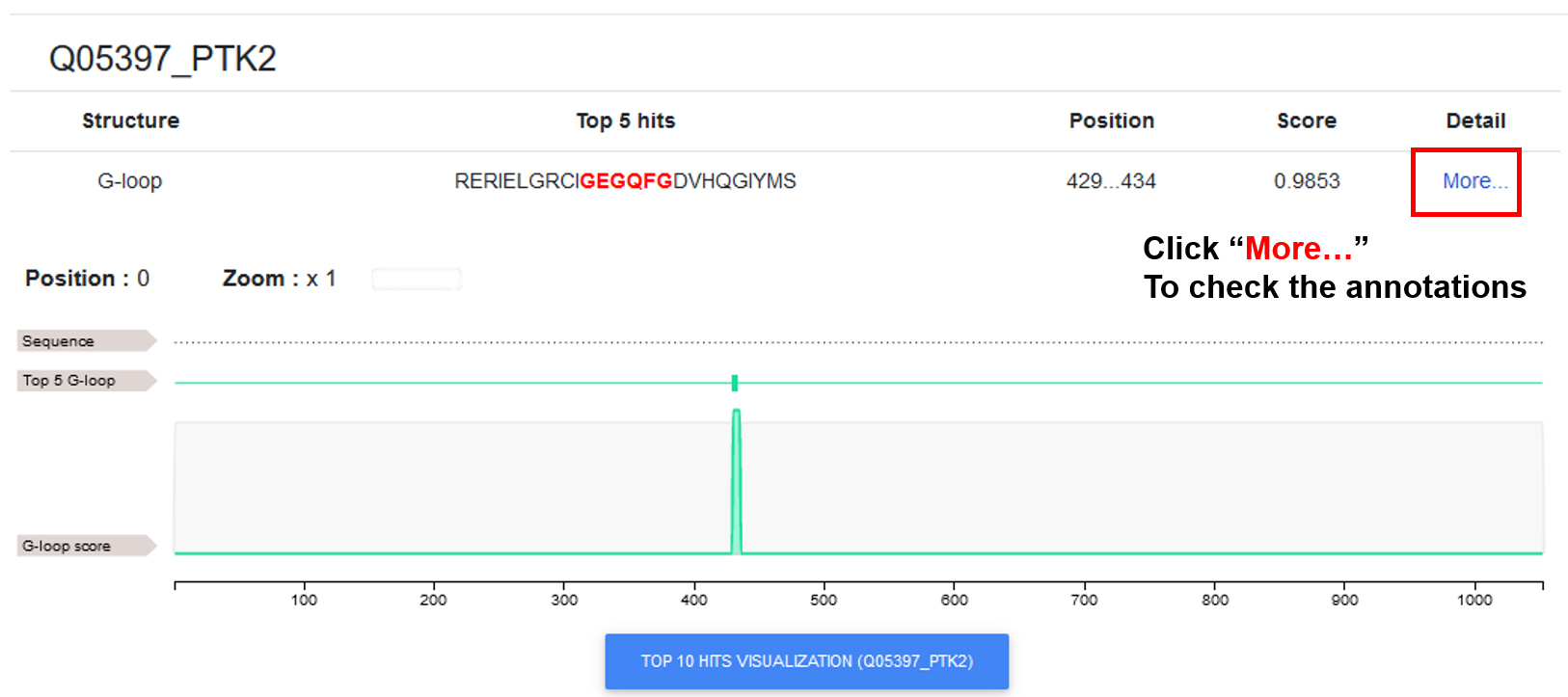
♦ The basic prediction information for the protein(s) is shown in this tab, including the DR hotspot type, DR hotspot instance, position, predicted score, and "Detail" link. By clicking on the different type buttons, users can view the results of the corresponding DR type. A line chart is added to visualize all the predictions, with the top 5 positions with higher score marked.
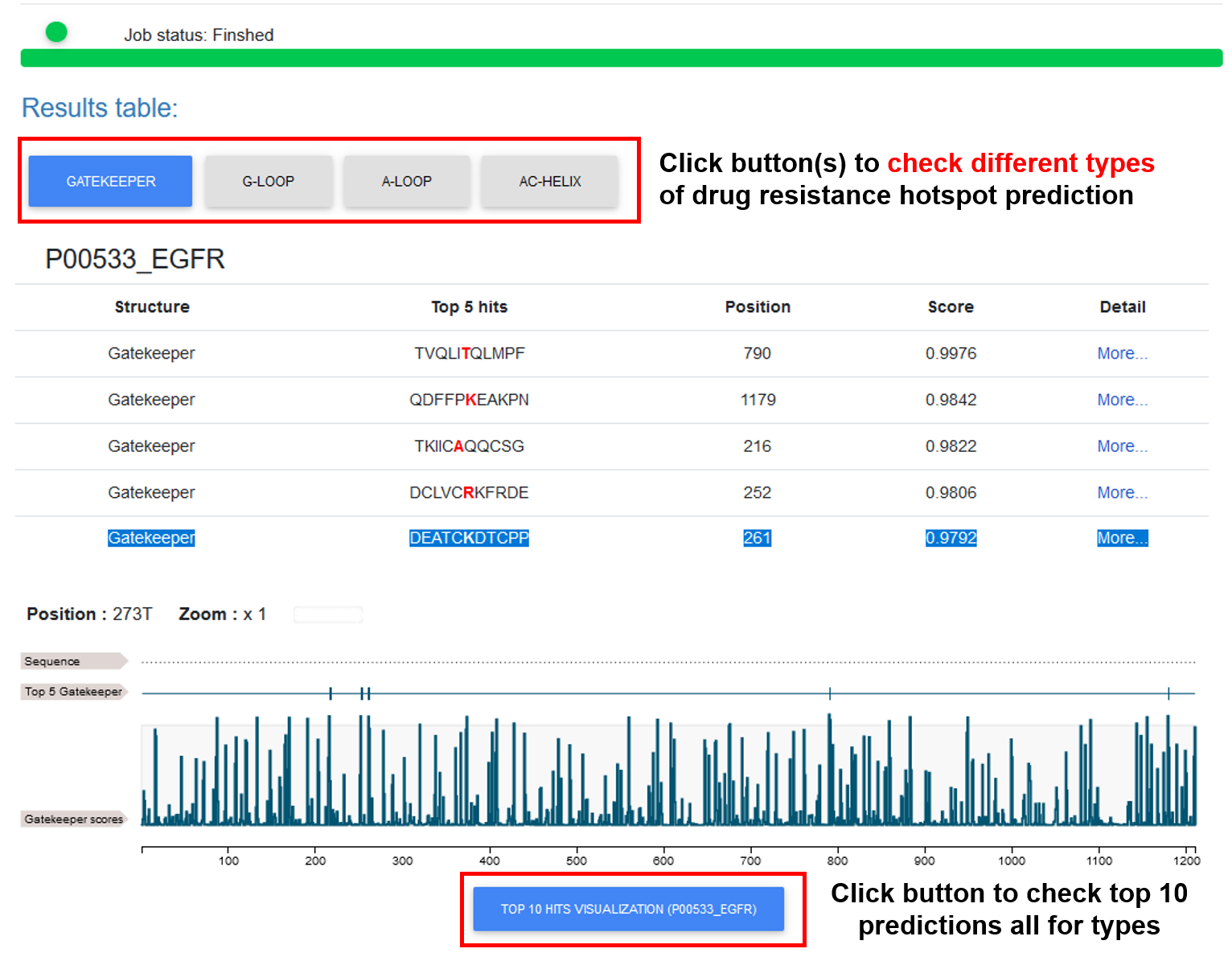
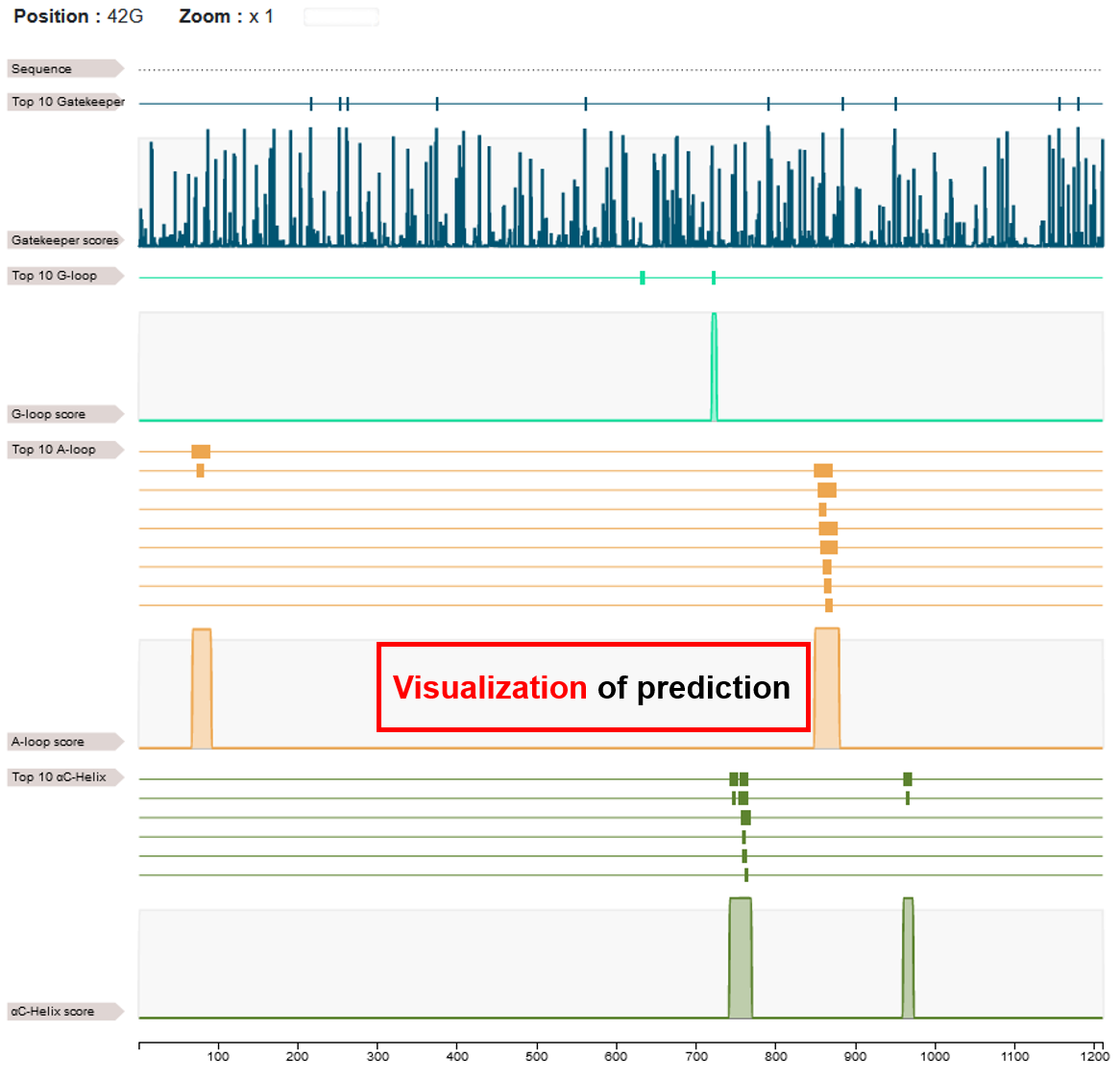
♦ By clicking on the "TOP 10 hit..." button, users can view the results of all predictions across selected DR types for the input protein. Users can select an area to zoom in for detailed annotations.
♦ By clicking on the "More..." link, users can view the comprehensive annotations for the prediction.
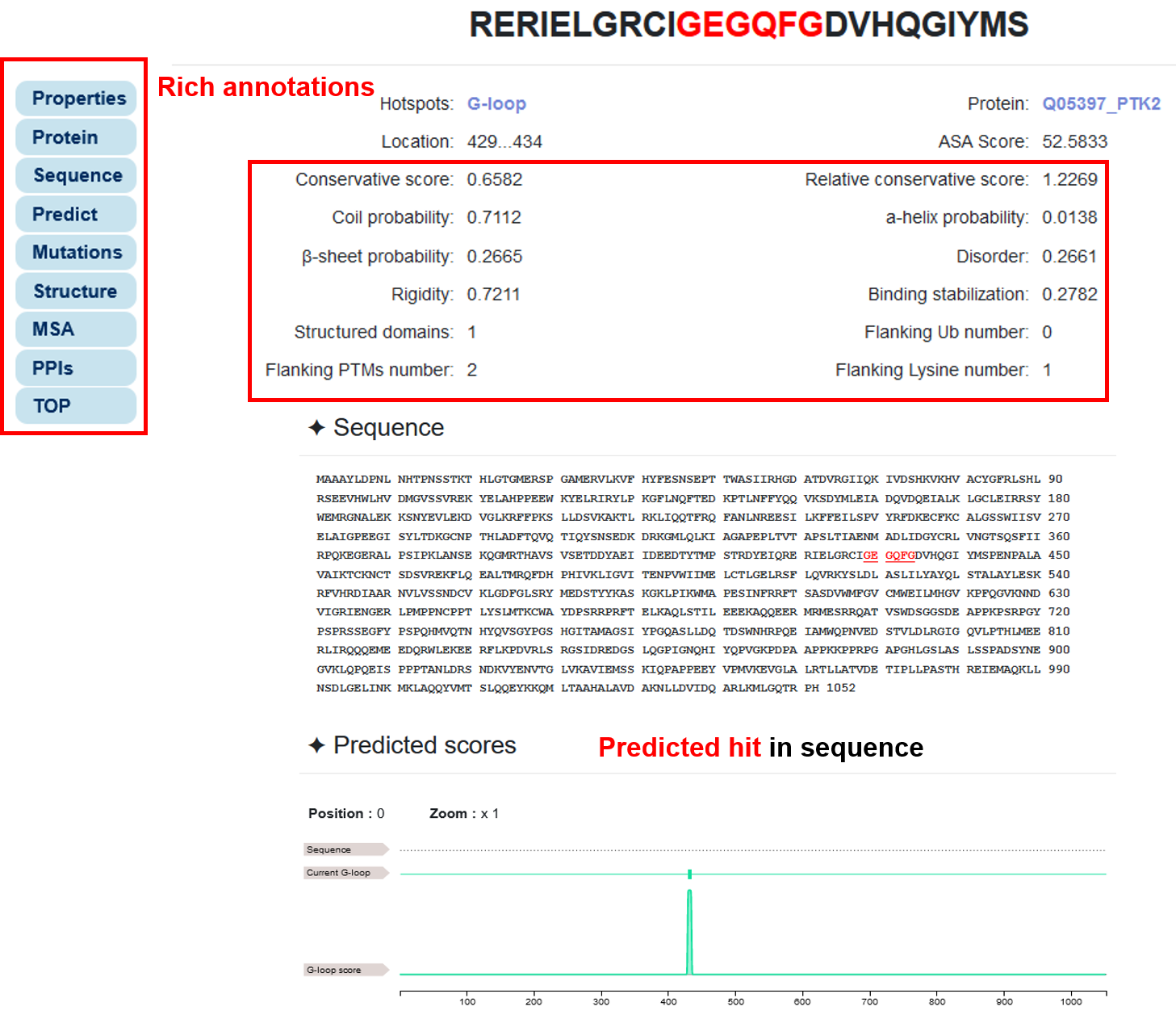
3.2 Rich annotations and visualizations
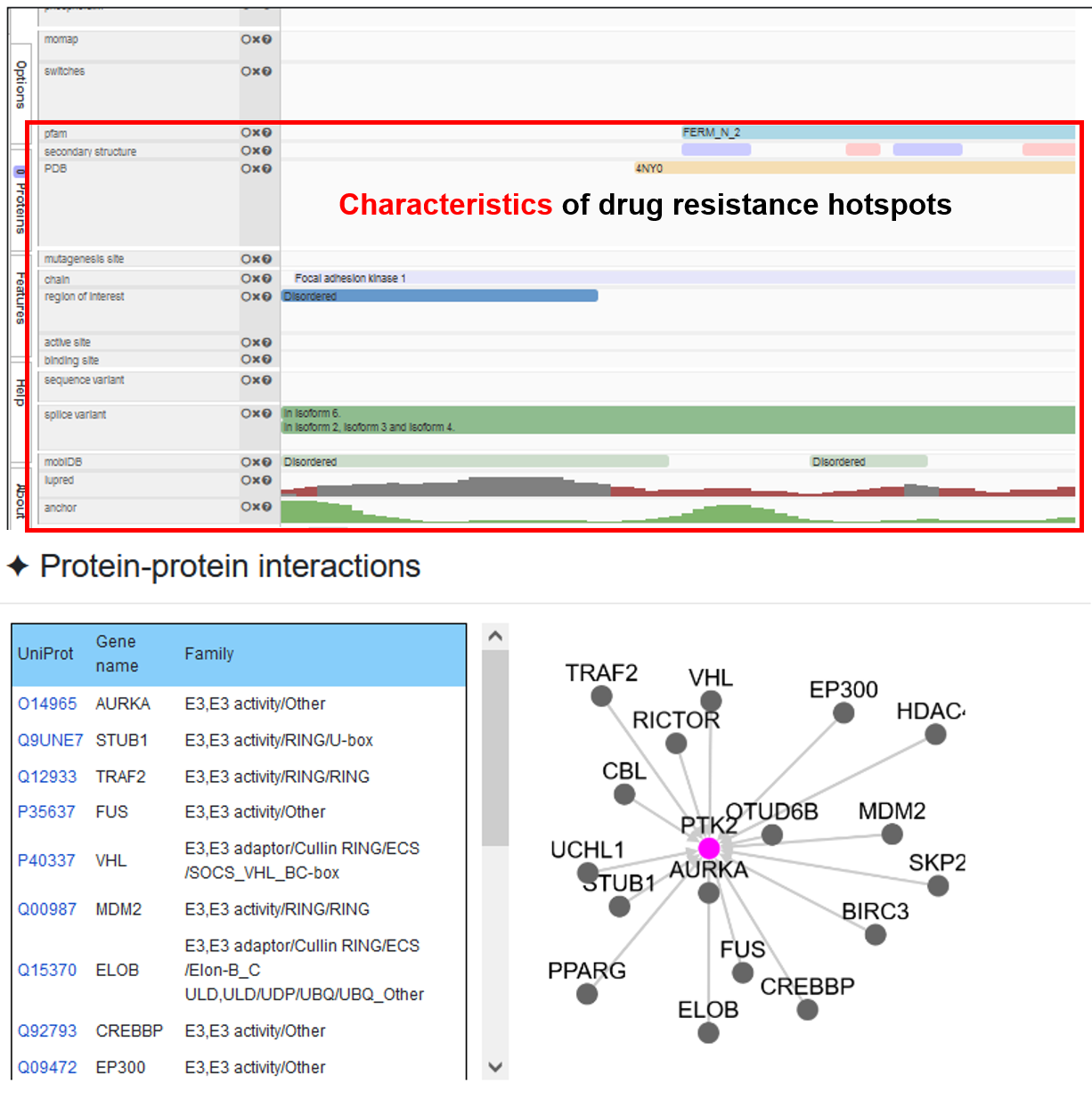
♦ In addition to the most basic predictions, we added a lot of annotation information and visualize it, including physicochemical properties, structural properties and conservation scores, source protein information, cancer mutations, 3D structure, multiple sequence alignment, and kinase-substrate network.
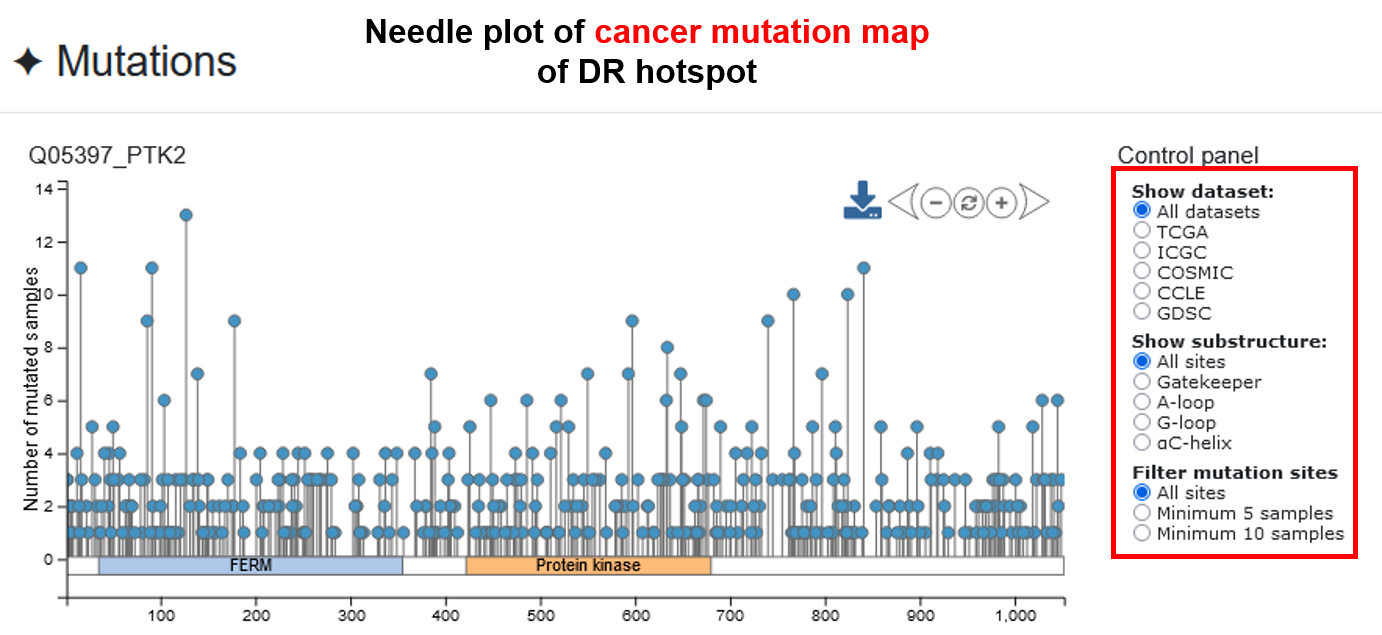
♦ A needle plot representing cancer mutation map in five genomics datasets was added. A bar plot displayed the number of mutated samples in each substructure. Clicking on the bar will show the number of mutated samples in each pan-cancer dataset.
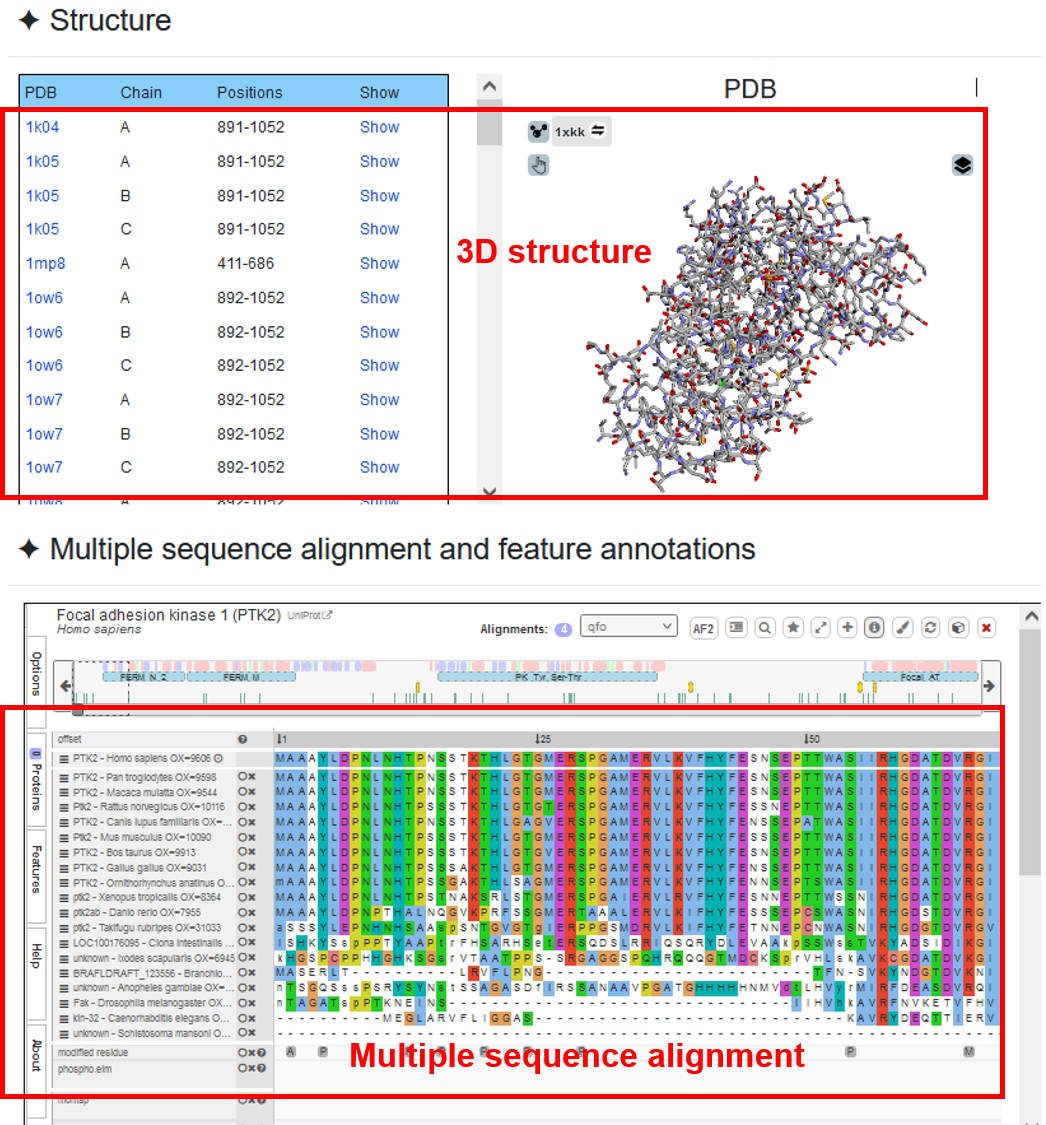
♦ The structure information of the query protein is mapped from PDB database and visualized, so that users can check if the DR hotspot is located on the important structural region. In addition, we processed and visualized the information of multiple sequence alignment to view the sequence conservation of DR hotspots.
♦ Information on physicochemical properties, structural properties and functional domains predicted by multiple bioinformatics tools is supplemented and visualized as well as substrate information.
4. How to cite
♦ Lin, S, Tu, C, Hu, R, Wang, H, Dong, Z, Luo, H, Kuang, L, Wang, T, Wang, L, Zhao, Z, Li, Z, & Xu, H. (2025). Dr. Kinase: predicting the drug-resistance hotspots of protein kinases. Nucleic Acids Research, 53(W1), W258–W265. [PMID: 40308214]
♦ Hu R, Xu H, Jia P, Zhao Z (2021). KinaseMD: kinase mutations and drug response database. Nucleic Acids Research, 49 (D1), D552-D561. [PMID: 33137204]
♦ Kim P, Li H, Wang J, & Zhao Z (2021). Landscape of drug-resistance mutations in kinase regulatory hotspots. Briefings in Bioinformatics, 22(3), bbaa108. [PMID: 32510566]